chinês
chinês

Vantagens da prótese total digital
2024-12-31
2026-07-06
Se você fabrica, distribui ou compra dispositivos médicos na Europa, provavelmente já se deparou com os termos MDD e MDR . Embora ambos se refiram à marcação CE para dispositivos médicos, representam dois quadros regulamentares diferentes com requisitos significativamente diferentes.
Desde o Regulamento de Dispositivos Médicos (UE) 2017/745 (MDR) tornou-se plenamente aplicável em 26 de maio de 2021 , gradualmente substituiu o antigo Diretiva de Dispositivos Médicos (93/42/EEC, MDD) . A transição introduziu requisitos mais rigorosos em matéria de provas clínicas, rastreabilidade de produtos, vigilância pós-comercialização e documentação técnica.
Para os fabricantes de materiais e equipamentos CAD/CAM odontológicos, compreender essas diferenças é essencial para manter a conformidade e acessar o mercado europeu.
O Diretiva de Dispositivos Médicos (93/42/CEE) foi introduzido pela União Europeia em 1993 para harmonizar as regulamentações de dispositivos médicos em todos os estados membros.
Como um Diretiva , o MDD estabeleceu requisitos gerais que cada estado membro da UE incorporou na sua própria legislação nacional. Embora esta consistência tenha melhorado, a implementação pode variar ligeiramente entre os países.
Sob o MDD, os fabricantes eram obrigados a:
Durante muitos anos, o MDD serviu como principal quadro regulamentar para dispositivos médicos vendidos na Europa.
O Regulamento de Dispositivos Médicos (UE) 2017/745 , comumente conhecido como MDR, foi adotado em 2017 e tornou-se totalmente aplicável em 26 de maio de 2021 .
Ao contrário do MDD, o MDR é um Regulamento , o que significa que se aplica direta e uniformemente em todos os estados membros da UE, sem exigir implementação nacional.
Os objetivos principais do MDR incluem:
O MDR estabelece uma abordagem de ciclo de vida mais abrangente, exigindo que os fabricantes monitorem o desempenho do produto mesmo após a obtenção da certificação CE.
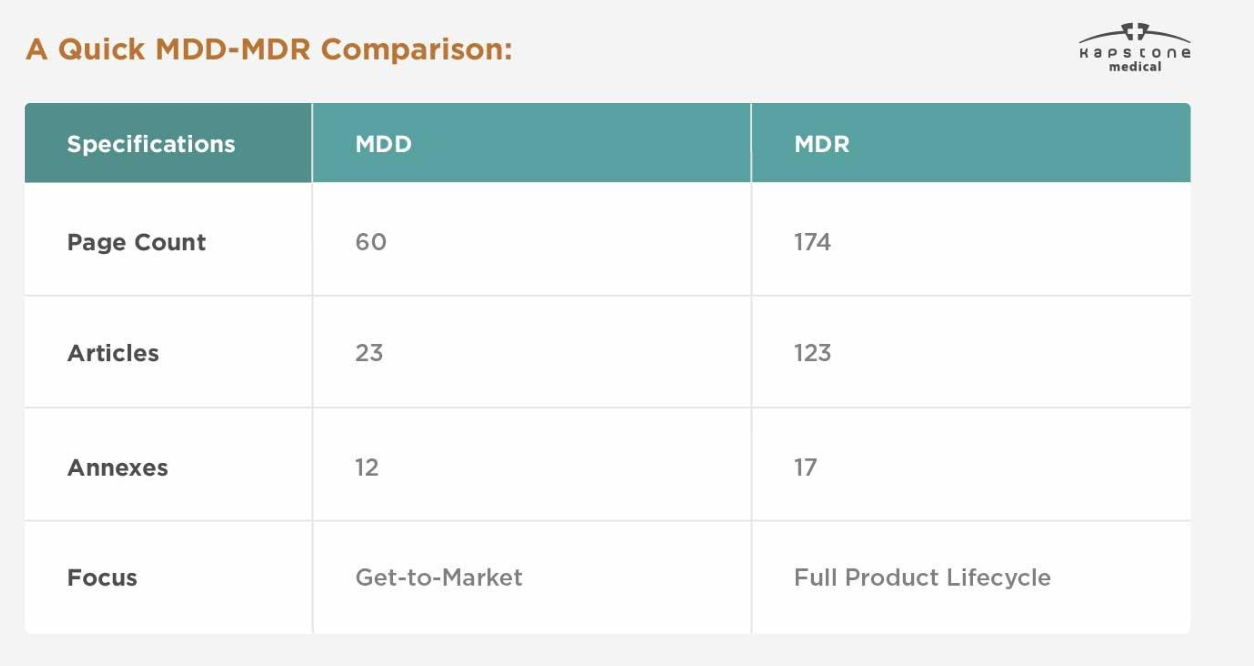
| Categoria | MDD | MDR |
|---|---|---|
| Quadro Legal | Diretiva | Regulamento |
| Referência da UE | 93/42/EEC | (UE) 2017/745 |
| Implementação | Legislação nacional | Diretamente aplicável em toda a UE |
| Evidência Clínica | Requisitos limitados | Requisitos significativamente aprimorados |
| Sistema UDI | Não obrigatório | Obrigatório para dispositivos aplicáveis |
| Registro EUDAMED | Não disponível | Obrigatório de acordo com os cronogramas de implementação |
| Vigilância Pós-Mercado | Básico | PMS e PMCF abrangentes |
| Documentação Técnica | Padrão | Mais detalhado e mantido continuamente |
| Supervisão Regulatória | Moderado | Muito mais rigoroso |
A mudança mais significativa é que o MDR exige que os fabricantes gerenciem a conformidade durante todo o ciclo de vida do produto, e não apenas antes da entrada no mercado.
A tecnologia médica evoluiu rapidamente nas últimas três décadas. A odontologia digital, o software assistido por IA, os biomateriais avançados e os dispositivos médicos conectados exigem uma supervisão regulamentar mais forte do que a estrutura original do MDD foi concebida para fornecer.
O MDR foi introduzido:
Estas melhorias ajudam a garantir que os dispositivos médicos continuem a cumprir os requisitos de segurança e desempenho após entrarem no mercado.
A resposta curta é não .
Novos dispositivos médicos não podem obter novos certificados MDD.
Os fabricantes que procuram a marcação CE para produtos que entram no mercado europeu devem cumprir os requisitos do MDR.
Alguns dispositivos que anteriormente possuíam certificados MDD válidos beneficiaram de disposições transitórias estabelecidas pela UE, desde que fossem cumpridas condições legais específicas. No entanto, estas disposições são temporárias e destinam-se a apoiar a transição para o MDR e não a substituí-lo.
Para empresas que planejam lançamentos de novos produtos, a certificação MDR é o caminho regulatório aplicável.
A indústria odontológica passou por uma transformação digital significativa na última década. Como resultado, muitos produtos utilizados em laboratórios dentários modernos enquadram-se no âmbito do MDR.
Exemplos incluem:
Os fabricantes devem demonstrar a consistência do material, a biocompatibilidade, o gerenciamento de riscos e o desempenho clínico por meio de documentação técnica abrangente.
Dependendo da finalidade médica e da classificação pretendidas, os fabricantes podem precisar fornecer evidências adicionais relacionadas à segurança, validação de software e gerenciamento de qualidade.
Os sistemas de digitalização digital geralmente incluem componentes de software que exigem documentação do ciclo de vida, considerações de segurança cibernética e processos de validação sob MDR.
Quando aplicável como dispositivos médicos, os fabricantes devem manter documentação técnica completa e implementar atividades contínuas de vigilância pós-comercialização.
Para os fabricantes de produtos odontológicos, o MDR representa não apenas um requisito regulatório, mas também uma oportunidade de demonstrar a qualidade do produto e a confiabilidade a longo prazo.
Comparado ao MDD, o MDR dá maior ênfase à conformidade contínua durante todo o ciclo de vida de um produto.
As principais adições incluem:
Esses requisitos ajudam a melhorar a rastreabilidade dos produtos, facilitam os recalls quando necessário e proporcionam maior confiança aos profissionais de saúde e aos pacientes.
Os certificados MDD existentes podem permanecer válidos apenas sob disposições transitórias específicas estabelecidas pela União Europeia. Os fabricantes devem verificar se os seus produtos se qualificam e monitorar os prazos aplicáveis.
Não. As novas certificações CE para dispositivos médicos destinados ao mercado da UE devem seguir os requisitos do MDR.
Sim. O MDR é o principal quadro regulamentar que rege a maioria dos dispositivos médicos colocados no mercado da UE.
Muitos materiais odontológicos, sistemas CAD/CAM, scanners e outros dispositivos médicos se enquadram no escopo do MDR, dependendo da finalidade e classificação pretendidas.

O MDD e o MDR partilham o mesmo objectivo – garantir que os dispositivos médicos colocados no mercado europeu sejam seguros e eficazes – mas diferem significativamente tanto na estrutura como nas expectativas regulamentares.
Embora o MDD tenha estabelecido a estrutura original para a certificação CE, o MDR introduz uma abordagem de ciclo de vida muito mais abrangente, enfatizando evidências clínicas mais fortes, rastreabilidade aprimorada, vigilância pós-comercialização aprimorada e maior transparência.
Para fabricantes de materiais e equipamentos CAD/CAM odontológicos, compreender o MDR não é mais opcional. É uma parte essencial da conformidade regulamentar, do acesso ao mercado internacional e da competitividade empresarial a longo prazo.
Ao investir em sistemas robustos de gestão de qualidade, documentação técnica abrangente e conformidade regulatória contínua, os fabricantes podem atender melhor às expectativas em evolução da indústria, ao mesmo tempo que fornecem produtos mais seguros e confiáveis aos profissionais de odontologia em todo o mundo.

Fresamento a seco e úmido para zircônia, PMMA, cera com trocador automático de ferramentas.
saber mais
Digitalização 3D de alta precisão, calibração AI, precisão de arco completo.
saber mais
Sinterização completa em 40 minutos com 57% de translucidez incisal e resistência de 1050 MPa.
saber mais
Scanner ultrarrápido com precisão de 5 mícrons e exportação STL aberta.
saber mais
Ciclo de 40 min para 60 coroas, cadinho de dupla camada e aquecimento de 200°C/min.
saber mais
Impressora LCD de alta velocidade para guias, temporários, modelos com resolução 8K.
saber mais